26
Bifar
Sección
Científica
Enfermedad
de GAUCHER.
Avances terapéuticos
a enfermedad de Gaucher, des-
crita por primera vez en 1882,
es la más frecuente de las enfer-
medades de depósito lisoso-
mal, con una frecuencia aproximada de
1:50.000 habitantes. Se produce como
resultado de la deficiente actividad de
la enzima lisosomal beta-glucocerebro-
sidasa. El déficit de la enzima produce
el acúmulo de un complejo glucolipí-
dico, la glucosilceramida, en los órga-
nos más ricos en células del sistema
mononuclear–macrofágico preferente-
mente el hígado, el bazo o los huesos,
aunque en un menor número de casos
puede afectar también al sistema ner-
vioso central o los pulmones.
necrosis, infartos o fracturas, mien-
tras que en otras ocasiones a pesar
de haber heredado el mismo defecto
genético, los pacientes apenas tienen
una leve esplenomegalia o discreta
trombocitopenia asintomática.
Por otra parte, el compromiso que tiene
el macrófago con el sistema inmune,
implica que estos pacientes tengan
una mayor incidencia de neoplasias
sobre todo hematológicas como mie-
loma múltiple o neoplasias de células
B y otras enfermedades neurológicas
no consigue evitar todos los síntomas
de la enfermedad.
Actualmente hay varias terapias aplica-
bles en la EG, dirigidas a incrementar
la degradación de los complejos glu-
colipídicos mediante la infusión perió-
dica de una beta-glucocerebrosidasa
recombinante. Actualmente existen dos
enzimas disponibles en Europa, la pri-
mera obtenida en cultivos de células
CHO (Chinese Hamster Ovarium) (Imi-
glucerasa) y la segunda, más reciente,
comercializada desde 2011, obtenida
de cultivos de fibroblastos humanos
(Velaglucerasa), la secuencia de ami-
noácidos de esta última es idéntica a
la natural. Existe una tercera enzima
obtenida en células vegetales (Taliglu-
cerasa) autorizada para su administra-
ción en USA, Brasil, Israel y otros paí-
ses de Hispano-América.
Otra opción de tratamiento consiste en
disminuir la síntesis de glucosilcera-
mida inhibiendo a la enzima ceramida-
glucosiltransferasa con lo que se evita
la formación de sustrato; miglustat es
un tratamiento de administración oral
derivado de un azúcar que ha demos-
trado en los diez años de experiencia,
que es eficaz para controlar los sínto-
mas de la enfermedad en los pacientes
con formas leves o moderadas.
Otro inhibidor de substrato análogo de
la ceramida, de administración oral, y
que bloquea también a la enzima cera-
mida-glucosiltransferasa, se encuentra
todavía en fase experimental.
Las últimas novedades, destinadas a
mejorar la eficacia del tratamiento y
facilitar la administración del mismo,
están encaminadas a desarrollar líneas
de investigación en la aplicación de
pequeñas moléculas que ayudan a
recuperar la actividad residual de las
enzimas mutadas, se denominan cha-
peronas farmacológicas y constituyen
una promesa para realizar tratamiento
dirigido en pacientes con ciertas muta-
ciones susceptibles de responder a
este tipo de tratamiento para incremen-
tar la actividad enzimática.
Por último, en la 10ª Reunión del Sim-
posio WORLD en enfermedades liso-
somales de 2014, celebrado reciente-
mente en San Diego, se han presen-
tado los resultados del ensayo clínico
con la nueva formulación oral de la
enzima glucocerebrosidasa expresada
Blanca Medrano Engay.
Farmacéutica. Unidad de Investigación Traslacional (UIT).
Hospital Universitario Miguel Servet.
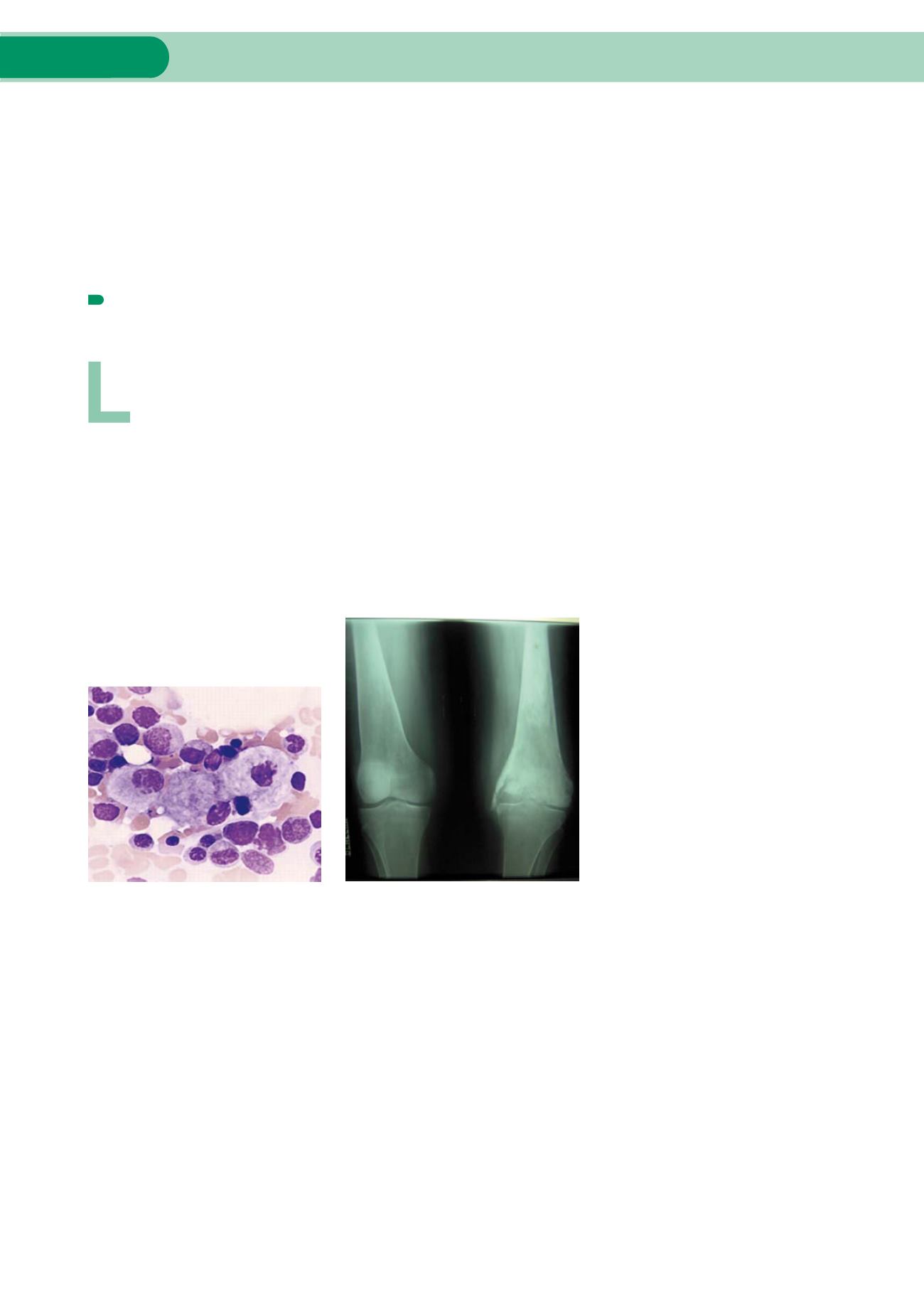
Radiografía de paciente con deformación a
nivel de rodillas
como la Enfermedad de Parkinson. La
alteración metabólica induce también
con frecuencia problemas hepatobilia-
res no resueltos.
Desde hace más de 20 años, los afec-
tados por esta enfermedad disponen
de un tratamiento eficaz: la enzima
sustitutiva, inicialmente obtenida de
placenta humana y más tarde sinteti-
zada por biotecnología en células de
mamífero, que administrada por vía
intravenosa de forma periódica, consi-
gue reducir los síntomas y evitar com-
plicaciones en un porcentaje elevado
de casos, sin embargo, el tratamiento
Es una enfermedad producida por una
alteración genética y por tanto heredi-
taria, pero aunque se nace con ella,
la diversidad clínica que la caracteriza
hace que pueda pasar desapercibida
hasta la edad adulta. El conocimiento
de la fisiopatología de la enfermedad
abre muchas líneas de investigación
tanto en los aspectos genéticos, como
en las diferentes manifestaciones clíni-
cas de la enfermedad. Se buscan fac-
tores que justifiquen el porqué en algu-
nos pacientes la enfermedad tiene un
comportamiento agresivo, provocando
intensos dolores en los huesos o com-
plicaciones irreversibles como osteo-
Células que acumulan glucocerebrósido:
células de Gaucher


