30
Bifar
clínicos juzgaron adecuado continuar usándo-
la contra las formas más complejas de tripa-
nosomiasis.
Alfred Bertheim, director
de Hoechst Dyeworks
,
sintetizó en el año 1907 un derivado del arse-
nofenol que era nominalmente el compuesto
606 de una serie de síntesis. Un análisis erróneo
llevó a considerarlo ineficaz como medicamen-
to tripanosomicida; y durante un año aproxima-
damente quedó olvidado como un compuesto
fallido.
Treponema pallidum
Treponema pallidum
, microorganismo etiológico
de la sífilis, fue descrito por
Fritz Schaudinn
y
Erich Hoffmann
, en Reichsgesundheitsamt, en
Berlín, en el año 1905.
Hoffmann propuso a Ehrlich estudiar sus com-
puestos arsenicales considerando la similitud
entre las espiroquetas causantes de la sífilis y
los tripanosomas responsables de la “enferme-
dad del sueño”. Hoffmann llevó a cabo ensayos
con estos compuestos en pacientes sifilíticos in-
gresados en su Clínica de Bonn, donde era res-
ponsable de Dermatología.
Al mismo tiempo, Paul Ehrlich suministró sus
productos a
Albert Neisser
, director a la sazón
de una Clínica Dermatológica en Breslau. Albert
Neisser llevó a cabo en el año 1907 estudios
en la isla indonesia de Java con Arsacetina y
Arsenofenilglicina. Los ensayos se realizaron en
simios a los que se había infectado con cepas
de
Treponema pallidum
. Estos estudios, publi-
cados en el año 1908, confirmaron indubitada-
mente la eficacia de Arsenofenilglicina. Esta fue
la primera vez que los compuestos sintetizados
por Ehrlich y sus colaboradores fueron ensaya-
dos en animales (recordemos que el único en-
sayo experimental en animales fue realizado con
arseniato sódico en roedores por Felix Nesmil).
A este ensayo le seguiría pronto el modelo de
infección sifilítica experimental en ratones, de-
sarrollado por Sacachiro Hata
en
Kitasato Ins-
titute
, en Tokio, Japón. Los estudios de Hata
recuperaron del olvido al denominado “com-
puesto 606”, dados los excelentes resultados
cuando se probaron en ratones a los que había
infectado de manera experimental con cepas de
Treponema pallidum
. Y así, el 10 de junio del
año 1909, Paul Ehrlich formuló una solicitud de
patente del “606” que fue presentada por
Far-
bwerke Hoechst
.
Muestras del “compuesto 606” fueron enviadas
a dos conspicuos clínicos rusos:
Iversen
, en-
tonces en el
Obuchow Hospital
para hombres,
en San Petersburgo; y a
Alt
, en
Uchtspringe
(Altmark), ambas ciudades en Rusia. Iversen
administró el compuesto a pacientes con infec-
ción causada por
Borrelia recurrens
, una infec-
ción transmitida por garrapatas. Se consideraba
que
Borrelia recurrens
y
Treponema pallidum
te-
nían gran semejanza microbiológica. Estos en-
sayos fueron paradigmáticos de otros que les
siguieron en distintos lugares y que confirmaron
la eficacia del “compuesto 606” frente a la sífilis.
10 de abril del año 1910: Paul Ehrlich presentó
su “compuesto 606” en el Congreso de Medici-
na Interna, en Weisbaden. El “compuesto 606”
fue bautizado como
Salvarsan
®, si bien era po-
pularmente conocido como “606” o “Ehrlich-
Hata 606”.
Muchos pacientes se consideraban curados
tras una sola inyección intramuscular conte-
niendo 900 mg. de Arsfenamina (
Salvarsán
®).
Arsfenamina en forma de polvo estaba conteni-
do en una ampolla sellada. Antes de la inyec-
ción se añadía un álcali para suspender el polvo
de Arsfenamina. La suspensión (sal sódica de
Arsfenamina) se debía inyectar rápidamente an-
tes de que precipitase de nuevo. Este proceder
resultaba relativamente complejo en algunos
casos. Poco después se adoptó un protocolo
algo más prudente, consistente en administrar
dosis más bajas (600 mg.), repitiendo la admi-
nistración a intervalos de varios días hasta una
dosis total acumulada de alrededor de 5 g. de
Arsfenamina.
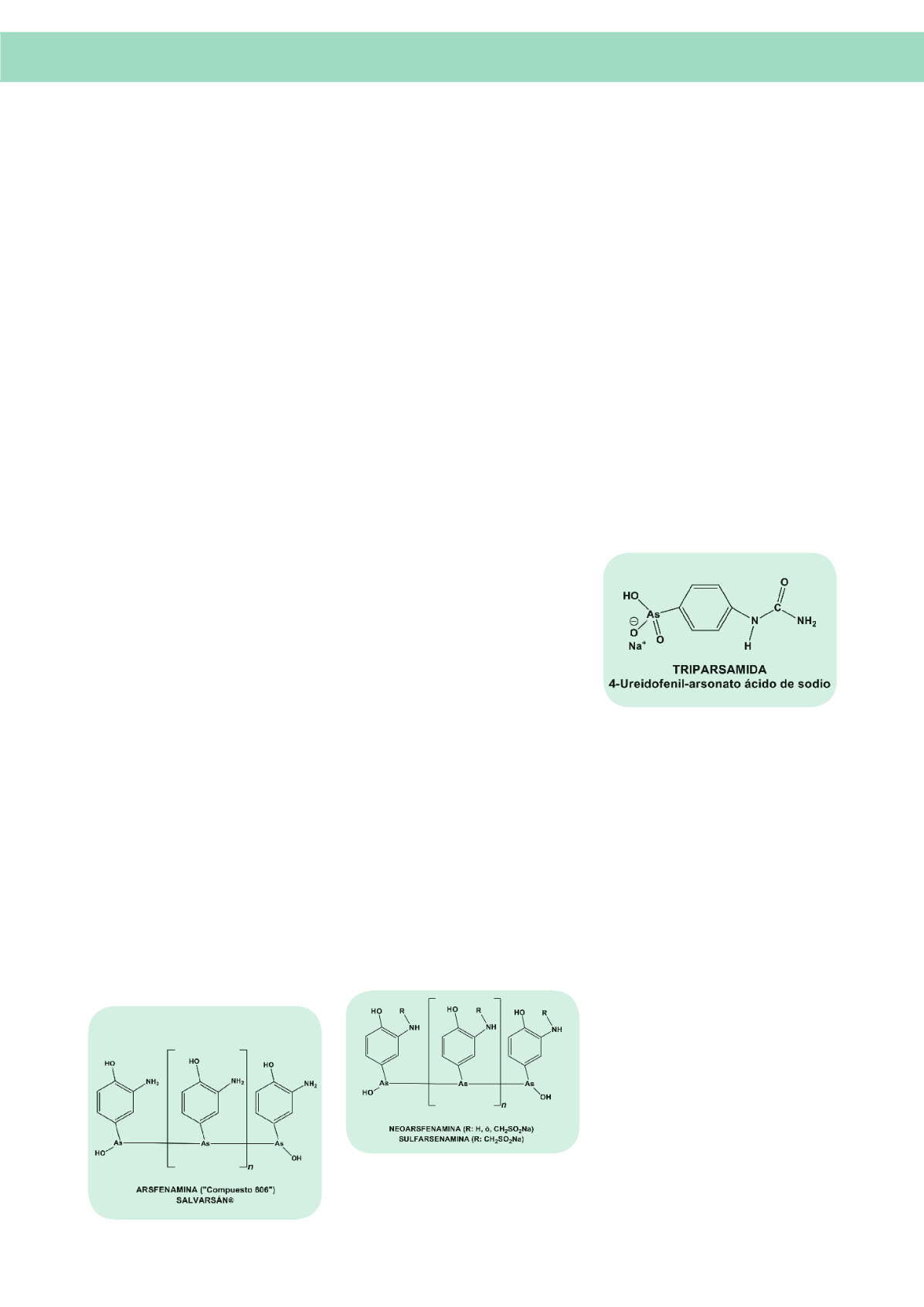
La Arsfenamina (
Salvarsán
®) y sus análogos
fueron el tratamiento estándar de la sífilis hasta
el final de la 2ª Guerra Mundial, cuando se dis-
puso de suficiente cantidad de Penicilina a un
coste asumible.
derivado hidrosoluble de la Arsfenamina. Pa-
ra ello Arsfenamina se trataba con sulfoxilato
de sodio (una sustancia usada en la industria
de tintes). Sulfoxilato sódico (CH
3
—SO
3
HNa)
se unía a algunos grupos amino del polímero
(Arsfenamina) haciendo el compuesto menos
susceptible a la oxidación. El compuesto así
formado se denominaba Neoarsfenamina (
Neo-
salvarsán
®). Aun cuando era algo menos eficaz
que Arsfenamina (
Salvarsán
®) tenía la ventaja
de una administración más fácil y cómoda. Esta
ventaja de índole práctica determinó que en muy
poco tiempo solo se usase Neoarsfenamina en
la praxis clínica. Este compuesto también fue el
único tratamiento del ántrax hasta el desarrollo
de los antibióticos.
Las inyecciones intramusculares de Neoarsfena-
mina eran muy dolorosas. La adición de un ex-
ceso de sulfoxilato sódico a la preparación daba
lugar a la formación de Sulfarsenamina, proce-
der que reducía de manera muy notable el dolor
asociado a la inyección intramuscular.
Muy poco tiempo después de iniciarse la pro-
ducción de Arsfenamina,
Hoechst Dyeworks
patentó Neoarsfenamina (
Neosalvarsán
®), un
En el año 1919,
Walter Jacobs
y
Michael Hei-
delberg,
del
Rockefeller Institute
de Nueva York,
introdujeron Triparsamida, un ácido arsónico de-
rivado de
Atoxyl
®. Triparsamida difundía en el
tejido nervioso y, en consecuencia, era eficaz
frente a la tripanosomiasis cerebral. Por des-
gracia, Triparsamida dañaba el nervio óptico,
al igual que
Atoxyl
®, compuesto del que deri-
va. No obstante continuó siendo un tratamiento
electivo contra la tripanosomiasis incluso bien
entrada la década de 1960; y hasta muy recien-
temente en las formas resistentes de tripanoso-
miasis (
vg
tripanosomiasis gambiana).
Rockefeller Foundation
patentó la Triparsamida.
Sin embargo renunció a las regalías del medi-
camento al objeto de facilitar el acceso de las
comunidades pobres donde la tripanosomiasis
cobra su peaje más elevado en costes de salud,
progreso y desarrollo. Esta actitud era acorde
con el espíritu presbiterano del magnate John
D. Rockefeller, quien creo la Fundación en el
año 1901.
Ernest Fourneau,
del Instituto Pasteur de París,
puso en entredicho que la
neurotoxicidad
de
los
ácidos fenilarsónicos
fuera consecuencia de las
elevadas dosis requeridas, sugiriendo que es-
taba relacionada con las impurezas de los pre-


